
Computational Materials Physics
We use computer simulations to study the properties of materials. By applying methods ranging from electronic structure calculations to classical molecular dynamics, we study liquids, colloids, and atomic or molecular crystals under different temperature and pressure conditions, both in and away from equilibrium.
Overview
Simulation is one of the major theoretical tools at our disposal to understand complex physical systems. Our research activities include the application of advanced computational methods to the study of materials in both the solid and liquid state. We use electronic structure methods such as density functional theory (DFT) as well as classical and ab initio molecular dynamics simulations. Apart from purely equilibrium problems (calculating partition functions or Schrödinger energy levels) we are also working on systems that are out of equilibrium. Examples include soft matter systems such as colloids subjected to shearing (with exotic phenomena such as jamming and rheological chaos); and mixed content fluids subjected to a steady chemical potential gradient (with complex phenomena that arise in drying, dissolution and mixing processes). In quantum-mechanical simulations of solid state phases we study the nonequilibrium dynamics of defects, such as grain boundaries; and use ab initio molecular dynamics of molecules at surfaces to address the kinetics of adsorption, desorption, and catalysis.
We have very strong links with in-house experimental programmes in Soft Matter Physics, in Quantum Ordering, and in Extreme Conditions Physics. We take every opportunity to confront our theories with the latest experimental data, and this makes for a very stimulating working environment for all concerned. The University of Edinburgh has led the UK in computational physics since that field was invented, and hosts the National Supercomputer, Archer.
Research interests
Below are some examples of the kinds of problems currently being addressed by our doctoral students and researchers.

Flow of Colloids and Fluid Mixtures
Colloids in suspension can exhibit new phenomena such as jamming and arrest, either spontaneously or driven by stress. These are being studied by adapting glass transition theories from statistical mechanics. We also have simulation work on the topic, mainly using a powerful algorithm called LB (Lattice Boltzmann). In future we will address the dynamics of colloidal particles suspended in fluid mixtures. These are capable of new forms of arrest, and should form viscoelastic solids at very low volume fraction. Figure 1 shows a binary fluid mixture undergoing demixing. The interface between fluids is shown blue on one side, yellow on the other; the fluids themselves are transparent.
Materials at Extreme Conditions
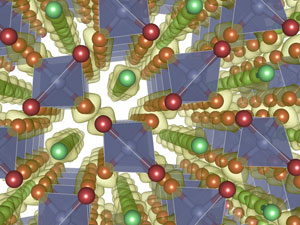
Under extremes of pressure, the behaviour of materials changes dramatically, often in ways that are hard to predict. Semiconductors such as Si and Ge become metals, while metals like Li, Na and K become insulators. Crystalline materials such as ice, Cs and Rb, can melt, while liquids can freeze. Magnetism and superconductivity can be lost, or appear. The promise of high-temperature superconductivity in compressed hydrogen is but one example. All these effects play a role in planetary sciences, materials science, and in our fundamental understanding of the interactions of matter in the condensed state.
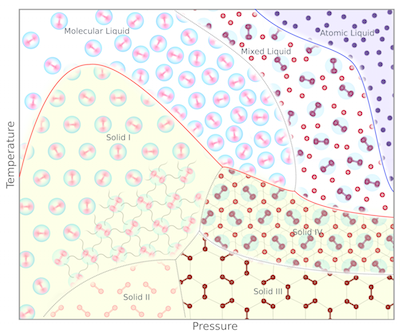
The basis for all these phenomena is quantum mechanics, and most can be understood using density functional theory, which translates the intractable problem of many interacting electron wavefunctions into a simpler problem involving the electron density. Solving the equations analytically is still beyond our means, but with several days of computing a reasonable description of any simple system at any pressure achievable in the lab can be obtained. Our calculations then serve to corroborate experimental data, explain why certain phenomena occur, or even predict new and exciting states of matter to motivate experimental studies.
Molecular Physics
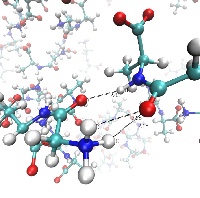
We are interested in understanding the relationship that exists between molecular (and electronic) structure and function. A complete elucidation and understanding of this relationship would be of great importance for molecular biology and medicine biotechnology, and other molecular-based technological devices. A particularly fruitful strategy is to examine small, relatively "simple" model systems, chosen such that, whilst being computationally tractable, they offer a sufficiently rich and diverse palette of physical phenomena that they allow an understanding of bigger, more complex systems. For example, small peptides can be viewed as fragments of larger proteins. We have a long-standing programme of research investigating such fragments using both classical atomistic and first principles quantum mechanical molecular dynamics methods, in collaboration with experimentalists at Rutherford Appleton Laboratory (RAL), and the National Physical Laboratory (NPL). esearch on such systems has provided valuable insights into the physical mechanisms by which peptides aggregate, whilst also clarifying how currently used classical potentials may be improved in order to better describe these systems.
A second, complementary strategy is to examine the behaviour of molecular crystals. Organic molecular crystals are capable of displaying a diverse range of physical phenomena, such as superconductivity, metal-insulator transitions, charge and spin ordering, Peierls transitions, etc. They thus provide an environment in which such physical phenomena may be probed theoretically and experimentally. From a technological perspective, such rich phase diagrams permit a crystal's properties to be "tuned", which may be used in, for example, sensing applications or molecular electronics. Density functional theory provides a powerful and flexible technique for the examination of such problems. Recent studies by the group have investigated pressure-induced metallisation in single-component molecular crystals, and modulating electronic properties through crystal polymorphism.
Two-dimensional van der Waals Materials
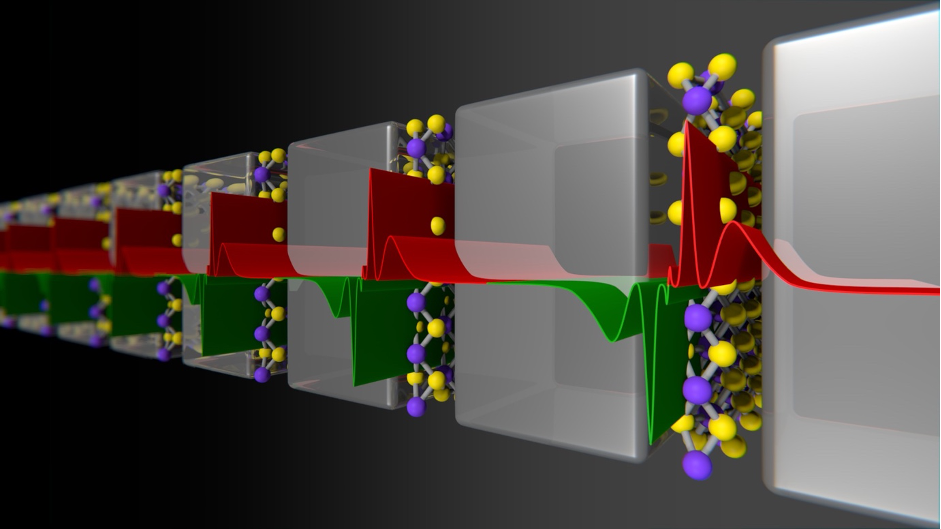
Two-dimensional (2D) van der Waals (vdW) atomic crystals, such as graphene and boron nitride have emerged as a new class of compounds with remarkable chemical and physical properties because of their unusual electronic structure. These thin, lightweight, bendable, highly rugged materials could bring dramatic changes to information processing and communications, with applications in conformal displays, wearable computers (e.g. Apple watch), and smart environments in human-electronic integration (e.g. touchscreens).
Among several 2D compounds, the realisation of long-range ferromagnetic order in vdW-layered materials has been elusive till very recently. Long searched but only now discovered 2D magnets are one of a select group of materials that retain or impart strongly spin correlated properties at the limit of few atoms thick. Such crystals are in high demand in several technological applications from sensing to data storage, which can lead to new magnetic, magnetoelectric and magneto-optic applications. However, it is not totally clear how such 2D materials could develop magnetism in few Å’s thickness. Indeed, what type of magnetic and topological order such 2D crystals can develop (e.g., ferromagnetic, antiferromagnetic, etc.) at such confined space is unknown. Here we explore, using multiscale computational methods (Monte Carlo, ab initio, spin Hamiltonians, Langevin dynamics, Landau-Lifshitz-Gilbert equation techniques), a broad variety of vdW compounds towards energy-efficient devices and applications. For instance, for information technologies (hard drives, data storage), sensors, quantum computing, etc.
Electronic and Thermoelectric Materials
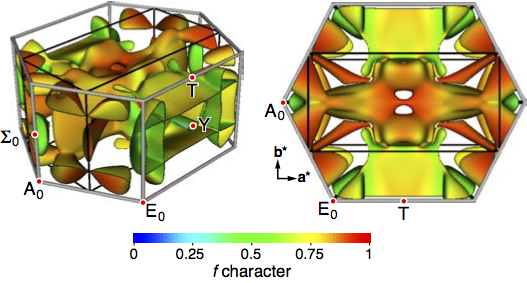
We want to understand the many-body effects in materials that lead to collective electronic phenomena such as superconductivity, exotic magnetism, multiferroicity, and topologically protected states. Those properties have the potential to revolutionise our electricity transport network, how we store digital information, and how we build new electronic devices. On the computational side, we can contribute by using parameter-free electronic structure methods to study the behaviour of the electrons in the respective materials; to understand the microscopic origin of the respective electronic effects; and to suggest to our experimental colleagues potentially interesting classes of materials, prior to their laboratory synthesis.
Thermoelectrics can convert heat directly to electricity if they are subjected to a temperature gradient. This offers the possibility to generate electrical power from the waste heat of, e.g., car engines, domestic boilers and industrial processes. However, the efficiency of presently available thermoelectric materials such as bismuth or lead telluride is not high enough and their cost is too high for general use beyond niche applications. The application of high pressure offers a means to tune the properties of thermoelectric materials, and substantial enhancements have been observed in a number of experiments. We use computational techniques to understand the physical mechanisms of these enhancements and to explore ways of recreating them without the application of pressure. The aim of this research is to help creating better-performing thermoelectrics for operation at normal pressure.
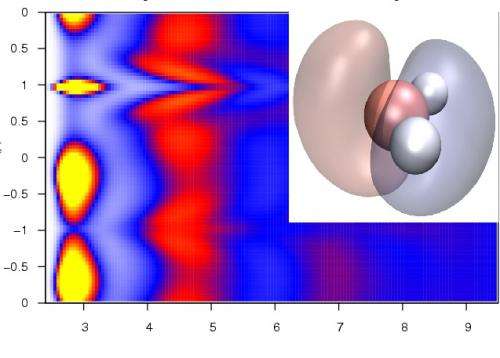
Method Development
In addition to the applied research highlighted above, we are constantly developing improved simulation methods. Our research in this area has a focus on the development and implementation of advanced sampling techniques for use in Monte Carlo and molecular dynamics simulations, such as lattice-switch methods; new methods of extracting experimental observables from simulation data, such as spectroscopic information; and the development of interatomic potentials, for instance for the description of intermetallic alloys, or polarisable water models for an improved description of liquid water in biomolecular simulations.
PhD project opportunities in Computational Materials Physics
- Active Self-Assembly of Colloidal Structures
- Active-Nematic/Polymeric Composites
- AI-Driven Discovery of Quantum Materials for Neuromorphic and Energy-Efficient Computing
- Alloys in the Earth's core
- Benchmarking DFT at extreme pressures
- Better Thermoelectric Materials Through High Pressure
- Computational Physics of Liquid Crystal Composites
- Is high pressure the end of hydrophobicity?
- Joint PhD studentship between the University of Edinburgh and the Institut Laue-Langevin (Grenoble)
- Purely Elastastic Instabilities and Turbulence
- Quantum simulations of rocky planet interiors
- Theory and Multiscale Simulations of DNA and Chromatin
- Topologically Active Polymers (Experimental and/or Computational)
- Transition to Turbulence
People in Computational Materials Physics
Telephone numbers in the list below are shown as UK numbers. Callers from outside the UK should remove the leading zero and use the UK country code (+44).
| Name | Position | Contact details | Location | Photo |
|---|---|---|---|---|
| Academic staff | ||||
| Graeme Ackland | Professor | gjackland [at] ed.ac.uk | JCMB 2602 |  |
| Chris Brackley | Lecturer in Theoretical/Computational Condensed Matter Physics | C.Brackley [at] ed.ac.uk | JCMB 2612 |  |
| Aidan Brown | Lecturer | Aidan.Brown [at] ed.ac.uk | JCMB 2616 |  |
| Andreas Hermann | Professor | a.hermann [at] ed.ac.uk | JCMB 2604 |  |
| Ingo Loa | Reader | I.Loa [at] ed.ac.uk | CSEC 3.3803 |  |
| Davide Marenduzzo | Personal Chair in Computational Biophysics | Davide.Marenduzzo [at] ed.ac.uk | JCMB 2506 |  |
| Miguel Martinez-Canales | Lecturer | Miguel.Martinez [at] ed.ac.uk | JCMB 2420 |  |
| Alexander Morozov | Professor | Alexander.Morozov [at] ed.ac.uk | JCMB 2507 |  |
| Miriam Pena Alvarez | UKRI Future Leaders Fellow | Miriam.Pena.Alvarez [at] ed.ac.uk | JCMB 3811 |  |
| Ciprian Pruteanu | Lecturer in Extreme Conditions Physics | cip.pruteanu [at] ed.ac.uk | JCMB 1608 |  |
| Elton Santos | Reader in Theoretical/Computational Condensed Matter Physics | esantos [at] ed.ac.uk | JCMB 2503 |  |
| Tyler Shendruk | Reader in Theoretical/Computational Condensed Matter Physics | t.shendruk [at] ed.ac.uk | JCMB 2422 | |
| Research staff | ||||
| Yair Augusto Gutierrez Fosado | Postdoctoral Research Associate in Biophysics | yair.fosado [at] ed.ac.uk | JCMB 1616 | |
| Marcin Kirsz | Postdoctoral Research Associate | marcin.kirsz [at] ed.ac.uk | JCMB 2607 |  |
| Davide Michieletto | Royal Society Research Fellow | Davide.Michieletto [at] ed.ac.uk | JCMB 1505 |  |
| Research postgraduates | ||||
| PeiYu Cai | Postgraduate Student | peiyu.cai [at] ed.ac.uk | JCMB 2501 | |
| Auro Varat Patnaik | Postgraduate Student | Auro.Patnaik [at] ed.ac.uk | JCMB 1511 | |
| Izzy Rodger | Postgraduate Student | izzy.rodger [at] ed.ac.uk | JCMB 3309 | |